Title here
Summary here
March 25, 20196 minutes
Solving Median String Problem in DNA using MapReduce

Genomes are fascinating. They are the sole source of information in living beings. Computational genetics, biochemistry and biology are rife with problems that are np-hard. As computer scientists, our goal is to come up with solutions that solve the problem biologists are facing in a way that is time and memory efficient.
So, what exactly are motifs? Before answering the question, let’s look how gene sequences look like. Wikipedia states that, “A nucleic acid sequence is a succession of letters that indicate the order of nucleotides forming alleles within a DNA (using GACT) or RNA (GACU) molecule. By convention, sequences are usually presented from the 5’ end to the 3’ end. For DNA, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.”
A gene sequence looks as shown below:
ACAAGATGCCATTGTCCCCCGGCCTCCTGCTGCTGCTGCTCTCCGGGGCCACGGCCACCGCTGCCCTGCC
CCTGGAGGGTGGCCCCACCGGCCGAGACAGCGAGCATATGCAGGAAGCGGCAGGAATAAGGAAAAGCAGC
CTCCTGACTTTCCTCGCTTGGTGGTTTGAGTGGACCTCCCAGGCCAGTGCCGGGCCCCTCATAGGAGAGG
AAGCTCGGGAGGTGGCCAGGCGGCAGGAAGGCGCACCCCCCCAGCAATCCGCGCGCCGGGACAGAATGCC
CTGCAGGAACTTCTTCTGGAAGACCTTCTCCTCCTGCAAATAAAACCTCACCCATGAATGCTCACGCAAG
TTTAATTACAGACCTGAAThe letters A, C, G and T are representative of the Nucleotides.
In genetics, a sequence motif is a nucleotide or amino-acid sequence pattern that is widespread and has, or is conjectured to have, a biological significance. For proteins, a sequence motif is distinguished from a structural motif, a motif formed by the three-dimensional arrangement of amino acids which may not be adjacent. Obviously, there can be mutations in the motif and the consensus motif can be different from the actual motif pattern in the DNA sequence.
Say, we have a long DNA sequence, how should we go about finding the consensus motif? Obviously, we have to have a predefined length for the motif. This is also called L-mer. For example, if you take target motif sequences of length 8, the target is to find 8-mer. Median String algorithm is one of the algorithms to find consensus motif. The algorithm is presented as follows:
Few Terms before we start the actual algorithm
$t$: Number of sample DNA sequences
$n$: Length of each DNA sequence
$DNA$: Sample of DNA sequences $t \times n$ array
$l$: length of the motif $ l - $ mer
$s_i$: Starting position of an $ l- $ mer
$s = (s_1, s_2, …, s_t) $: array of motif’s starting positions
An example is shown in the figure below:
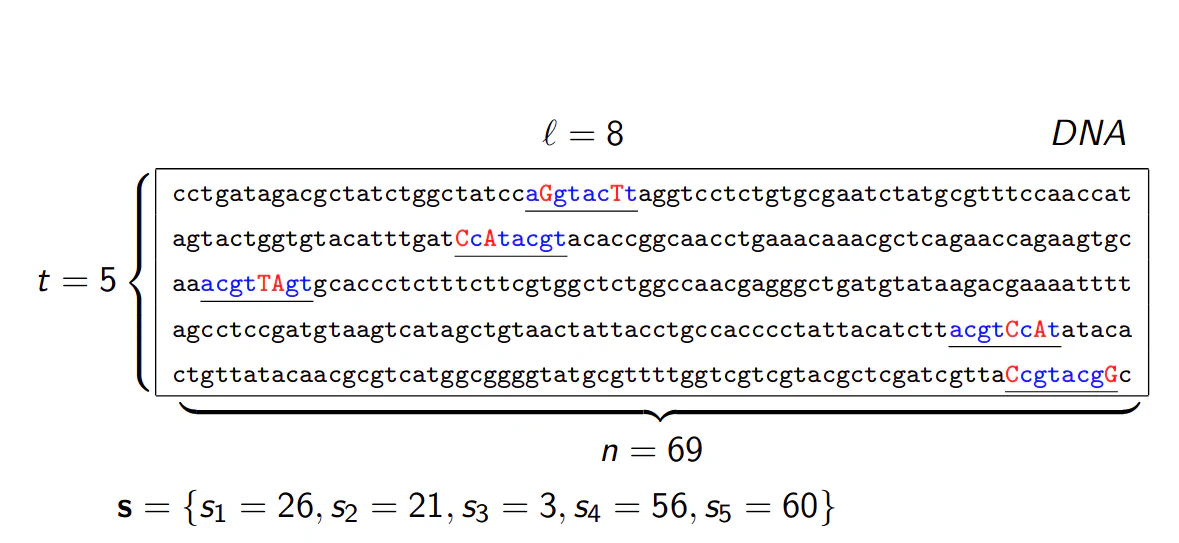
Given, $$s = (s_1, s_2, …, s_t) $$ and $$ DNA $$:
Here, $$ \text{count}(c, j) $$ gives the number of times that symbol c equals $$ DNA[i][s_i + j - 1] $$
The median string problem is formulated as:
The total distance is shown in the example below:
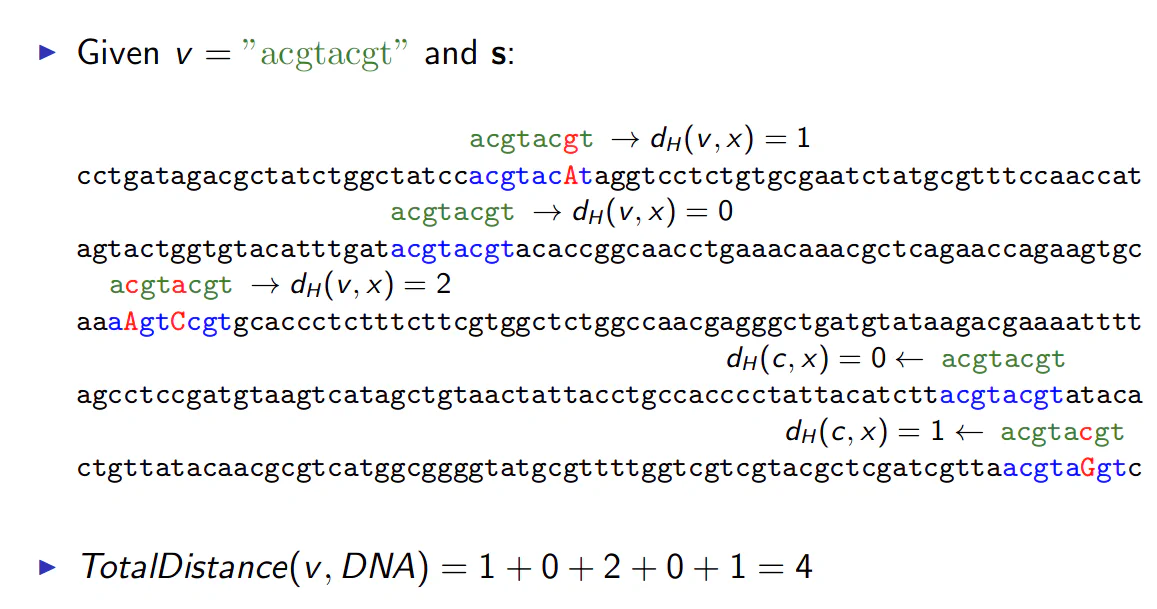
The median string problem is formulated as:
The algorithm is shown below:
$$ MedianStringSearch(DNA, t, n, l): $$
1: $ bestWord \longleftarrow AAA…A $
2: $ bestDistance \longleftarrow \infty $
3: $ \mathbf{for} $ each $l-mer$ $v$ from $AAA…A$ to $TTT…T$ $\mathbf{do}$:
4: $ \mathbf{if} $ $ TotalDistance(v, DNA) < bestDistance $ $ \mathbf{then}$:
5: $ bestDistance \longleftarrow TotalDistance(v, DNA) $
6: $ bestWord \longleftarrow TotalDistance(v, DNA) $
7: $ \mathbf{return }$ $ bestWord $
Now, consider the median string algorithm to find $ 8-mer $$. The total number of candidate target motifs from $$AAAAAAAA$$ to $$TTTTTTTT$$ is $$4^8$$. Let us convert the algorithm into hadoop map-reduce problem, as shown in the figure below:
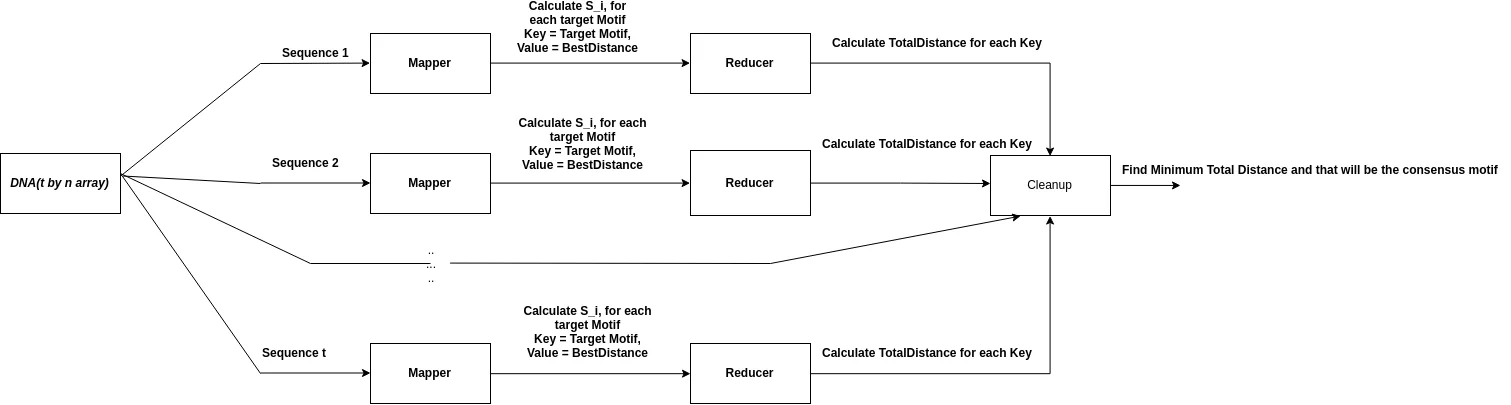
If you have a long DNA array spread over many lines and multiple text files, first thing you need to focus on is the way to parallelize the algorithm. One way to implement the algorithm with Hadoop is to do this:
<key, value> for the map operation output. Think of the target motif as the key and there will be multiple values for each key will be the best distance for each DNA seqeunce line.<key, values> : (targetmotif, bestDistancesForEachSequence) produced by the mapper and calculates the distanceScore for each key like this:Writable or WritableComparable interface. For example, if you want to send the sequenceId(line number), together with the best distance for each target motif from the mapper, you have to write your own class.In couple of days, I will be posting a link to a github repository that has the solution. I will be also posting instructions on how to run the code in Amazon EMR.